
Thesis opportunities of the Laboratory of Molecular Biology
The Laboratory of Molecular Biology offers a broad range of thesis projects for BSc and MSc students which are embedded in ongoing state-of-the-art research lines. If you see a topic that draws your interest, you can contact the group leader of the project directly to discuss options.

Plant architecture and development (Van Esse Group)
My group’s research focuses on understanding the genetic and molecular networks that are control different yield related traits such as developmental timing, seed and tiller number. We use natural and induced variation to study the genetic loci that control these key traits and state of the art ~omics tools to place these genes in precise molecular and cellular context.

Nodulation Engineering (Geurts Group)
The above title is a citation of a Science paper in 2016 (Stockstad, 2016, Science 353:12225-7) that reflected the massive expectations researchers have about engineering nitrogen-fixing root nodules on crops. Such root nodules are well known from legumes and are the subject of study for over a hundred years.
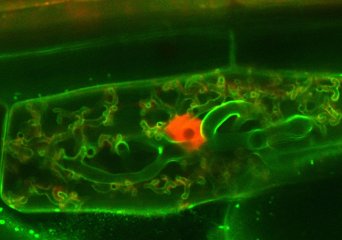
Molecular development of Arbuscular Mycorrhizal symbiosis (Limpens Group)
To live in environments where nutrients are limited, plants engage in an endosymbiosis with arbuscular mycorrhizal (AM) fungi. These fungi colonize plant roots and are hosted inside root cortex cells, where highly branched hyphal structures called arbuscules are formed (Figure 1).
RNA Biology of Plant Embryos (Nodine Group)
MicroRNAs (miRNAs) are short non-coding RNAs that mediate target transcript repression in plants and animals. Although miRNAs are required throughout development, relatively little is known regarding their embryonic functions.

Plant Developmental Systems (PDS, Angenent Group)
Within the PDS group we are interested in how developmental processes are controlled by transcription factors and chromatin modifications. We aim to unravel transcriptional networks underlying various processes such as flowering time regulation, floral organ development, fruit formation and embryogenesis.

Molecular control of flowering and reproduction of plants (Immink Group)
Flowering induction marks the start of the reproduction cycle and the final reproductive success of plants strongly depends on the timing of this developmental phase transition. Plants integrate various environmental cues, such as daylength, light quality and temperature with endogenous signals to time flowering, seed set and seed germination.